Spinal muscular atrophy (SMA) is a rare and debilitating autosomal recessive neuromuscular disease characterised by motor neuron degeneration and loss of muscle strength.1,2
Share page
Share now
A CLINICAL OVERVIEW OF SMA
The age of onset determines the level of motor neuron degeneration.1,2 Cognitive ability does not appear to be impacted by spinal muscular atrophy.2
Clinical literature shows a wide range of the incidence and prevalence of spinal muscular atrophy:
In the United States, the estimated incidence of spinal muscular atrophy is 8.5 to 10.3 per 100,000 live births.2,3
In Europe, the annual incidence varies greatly by country and type.4
Global annual incidence per 100,000 live births ranged from 3.5 to 7.1 for type I, 1.0 to 5.3 for type II, and 1.5 to 4.6 for type III.4
The lower motor neurons, located in the spinal cord, are important cells involved in motor function in the central nervous system (CNS) 5
For illustrative purposes only.
Share now
GENETICS
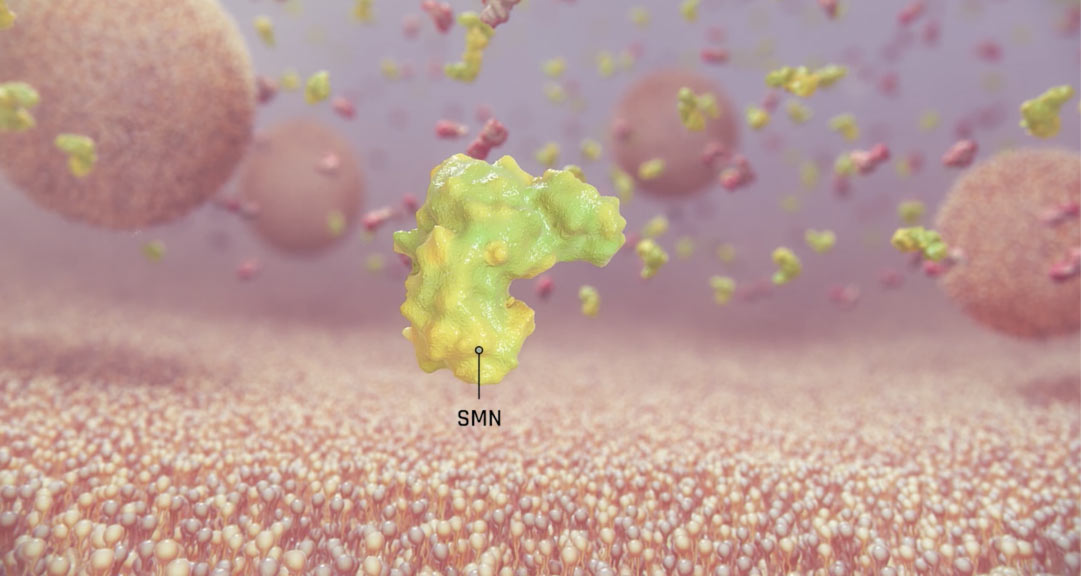
The genetic deficit underlying spinal muscular atrophy is well characterised. The role of the survival motor neuron 1 (SMN1) gene is to produce SMN protein, which is highly expressed in the spinal cord and is known to be essential for motor neuron survival.1,6
In SMA, homozygous mutations or deletions of SMN1 produce a shortage of SMN protein, which causes degeneration of motor neurons in the spinal cord.7,8
SMN1 gene6,7,9
The SMN1 gene is located on chromosome 5q13
Share now
SURVIVAL MOTOR NEURON 2
Nearly all people, including those with SMA, have a second, virtually duplicate gene to SMN1, known as survival motor neuron 2 (SMN2).2,10
SMN2 is nearly identical in genomic sequence to SMN1, with only five nucleotide differences7
However, a C-to-T nucleotide difference at position 6 of SMN2 creates an exonic splicing suppressor (ESS) that leads to a skipping of exon 7 during transcription2
This results in SMN2 producing a truncated, non-functional, and rapidly degrading SMN protein2
SMN2 gene 2,9
Share now
Approximately 10% of SMN2 transcripts result in full-length SMN protein, providing patients with an insufficient amount of SMN protein to sustain survival of spinal motor neurons in the CNS.2
SURVIVAL OF MOTOR NEURON PROTEIN SMN
Due to deficiency of the gene SMN1... 1,11
Two copies
Both copies are deleted or damaged
Partially compensated by the gene SMN2… 1,11
Copy number is variable
Only 10% SMN is functional
Share now
THE NUMBER OF SMN2 GENE COPIES IS INVERSELY RELATED TO THE SEVERITY OF SMA
Copy number of SMN2 is variable in patients with spinal muscular atrophy, and higher copy numbers of SMN2 correlate with less-severe disease:2
1 COPY
MORE THAN
95%
of individuals with spinal muscular atrophy retain at least 1 copy of SMN2
The copy number of SMN2 correlates to disease phenotype.
1 or 2 COPIES
ABOUT
80%
of individuals with Type I spinal muscular atrophy have 1 or 2 copies of SMN2
3 COPIES
ABOUT
82%
of individuals with Type II spinal muscular atrophy have 3 copies of SMN2
The higher the number of SMN2 gene the milder the disease type.
3 or 4 COPIES
ABOUT
96%
of individuals with Type III spinal muscular atrophy have 3 or 4 copies of SMN2
SMN2 copy number is related to, but not predictive of, disease severity, and care decisions should not be made based on copy number alone 12,13
In any case of spinal muscular atrophy, SMN2 copy number is less predictive of prognosis than age of onset and functional abilities14,15
In addition to SMN2, there is some evidence of other genetic modifiers of disease severity, including levels of the protein Plastin-313
Share now
Because of its potential role in modulating disease severity, SMN2 is a target for therapeutic interventions.16
The characters shown are real patients and the required consent to use their stories has been obtained from the patients and families. Photographs are for illustrative purposes only.
References
1. Lunn MR, Wang CH. Spinal muscular atrophy. Lancet 2008;371(9630):2120-2133.
2. Darras BT, Royden Jones H Jr, Ryan MM, De Vivo DC, eds. Neuromuscular Disorders of Infancy, Childhood, and Adolescence: A Clinician’s Approach. 2nd ed. London, UK: Elsevier; 2015.
3. Lally C, et al. Indirect estimation of the prevalence of spinal muscular atrophy Type I, II, and III in the United States. Orphanet Journal of Rare Diseases 2007; 12(1): 175.
4. Jones C. PP09.1 – 2352: Systematic review of incidence and prevalence of spinal muscular atrophy (SMA). European Journal of Paediatric Neurology 2015;19(Supp 1): S64–S65.
5. Islander G. Anesthesia and spinal muscular atrophy. Paediatr Anaesth 2013;23(9):804-816.
7. Lefebvre S, Bürglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995;80(1):155-165.
8. Medline Plus. Genetics Home Reference. SMN1. [online] 2016 Apr 20 [cited 2020 Sept 30]. Available from: URL: https://ghr.nlm.nih.gov/gene/SMN1.
9. Ogino S, Wilson RB. Spinal muscular atrophy: molecular genetics and diagnostics. Expert Rev Mol Diagn 2004;4(1):15-29.
10. Swoboda KJ. Romancing the spliceosome to fight spinal muscular atrophy. N Engl J Med 2014;371(18):1752-1754.
11. Rossoll W, Bassell GJ. Spinal Muscular Atrophy and a Model for Survival of Motor Neuron Protein Function in Axonal Ribonucleoprotein Complexes. Results Probl Cell Differ 2009;48:289-326.
13. Butchbach ME. Copy number variations in the survival motor neuron genes: implications for spinal muscular atrophy and other neurodegenerative diseases. Front Mol Biosci 2016;3:7.
14. Prior TW, Krainer AR, Hua Y, et al. A positive modifier of spinal muscular atrophy in the SMN2 gene. Am J Hum Genet 2009;85(3):408-413.
15. Burnett BG, Crawford TO, Sumner CJ. Emerging treatment options for spinal muscular atrophy. Curr Treat Options Neurol 2009;11(2):90-101.
16. Monani UR. Spinal muscular atrophy: a deficiency in a ubiquitous protein; a motor neuron-specific disease. Neuron 2005;48(6):885-896.